Contamination Control Strategy (CCS)
For Sterile Pharmaceutical Manufacturing
1. Purpose
This document outlines the Contamination Control Strategy (CCS) implemented at pharma industries for sterile pharmaceutical manufacturing. The objective is to proactively identify, control, and monitor all potential sources of contamination (microbiological, particulate, chemical, and pyrogenic) in accordance with EU GMP Annex 1 (2022) and US 21 CFR Parts 210/211.
CCS full form in Contamination Control Strategy in Pharmaceuticals.
2. Scope & Objectives
The CCS applies to all sterile production activities, critical cleanrooms (Grades A and B), isolators, Restricted Access Barrier Systems (RABS), supporting utilities, raw materials, personnel, and equipment involved in aseptic processing and sterile product manufacturing within the facility.
- Identify all potential sources and routes of microbiological, particulate, and pyrogenic contamination.
- Establish and implement effective controls for contamination prevention and reduction.
- Define procedural and technical measures to maintain environmental and product sterility.
- Provide a risk-based, documented framework aligned with regulatory requirements (EU GMP Annex 1, 21 CFR Part 211).
- Manage contamination through monitoring, deviation investigation, and continuous improvement.
3. Definitions
- Contamination: Unintended introduction of impurities (chemical, microbial, or particulate) into a sterile product.
- CCS: An integrated, site-wide strategy that addresses the design, control, and monitoring of all contamination risks.
- Aseptic Processing: A manufacturing process where sterile product is filled and sealed in a controlled, sterile environment.
4. Regulatory References
- EU GMP Annex 1 – Manufacture of Sterile Medicinal Products (2022)
- 21 CFR Part 210/211 – cGMP for Finished Pharmaceuticals
- ISO 14644 Series – Cleanroom Standards
- ICH Q9 – Quality Risk Management
- ICH Q10 – Pharmaceutical Quality System
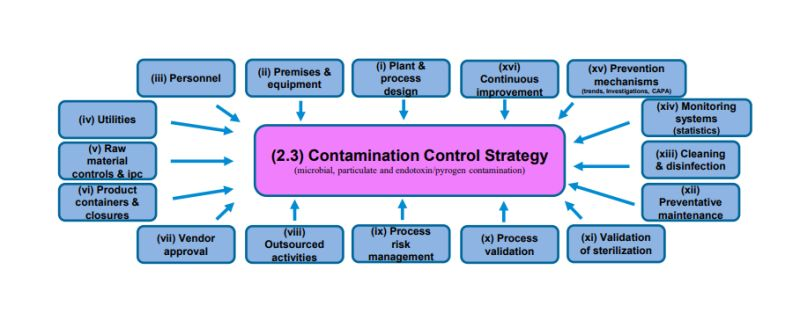
5. CCS Elements and Control Measures
5.1 Facility and Cleanroom Design
- Design cleanrooms to ensure unidirectional airflow, maintaining differential pressures preventing ingress of contaminants from less controlled zones. Designed using unidirectional flow principles with clear separation of Grade A, B, C, and D areas.
- Use smooth, non-shedding materials and validated cleaning/disinfection regimes for all surfaces and equipment.
- Ensure sterilization equipment and processes (including autoclaves, sterilizing filters) are qualified and validated.
- Implement robust maintenance programs minimizing contamination risk via planned servicing.
- Walls, ceilings, and floors made of non-shedding, easily cleanable materials.
- HVAC systems maintain pressure differentials and ISO-classified environments.
- Access control systems to prevent unauthorized entry.
5.2 Equipment Design and Qualification
- All equipment used in aseptic processing areas is qualified (DQ, IQ, OQ, PQ) and designed to minimize contamination risk (e.g., smooth surfaces, sealed gaskets).
- Use of closed or barrier systems (e.g., RABS, isolators) wherever possible.
- Regular cleaning and maintenance procedures validated and documented.
5.3 Personnel Control and Gowning
- Only trained, qualified personnel are allowed in cleanrooms.
- Initial and ongoing aseptic qualification, including media fill participation.
- Gowning is specific to each grade (A–D), validated for microbial containment.
- Personnel behavior monitored and reinforced through routine observation.
5.4 Process Design and Controls
- Critical parameters identified and controlled through validated processes.
- Use of sterile, depyrogenated components and containers.
- Sterilization processes (autoclaving, dry heat, filtration) are validated.
- In-process controls ensure integrity at critical steps.
5.5 Cleaning and Disinfection
- Cleaning agents and disinfectants validated for effectiveness against relevant microorganisms.
- Rotation of disinfectants to prevent resistance.
- Frequent and scheduled cleaning of all classified areas.
- Visual and microbiological checks post-cleaning.
5.6 Environmental Monitoring (EM) and Microbiological Monitoring
- Active and passive air monitoring for viable and non-viable particles.
- Surface monitoring (contact plates, swabs) on critical surfaces.
- Real-time alarms and alert/action limits for immediate investigation.
- Trending and data review integrated with CAPA systems.
- Maintain a comprehensive environmental monitoring program covering viable and non-viable particles in air, surfaces, personnel, and utilities per defined sampling plans.
- Use trending and risk assessment tools to evaluate monitoring data and trigger investigations for excursions.
- Validate and monitor utilities such as water-for-injection (WFI), compressed air, and HVAC systems to ensure contamination-free supply.
5.7 Utilities (Water, Air, Steam)
- WFI (Water for Injection): Loop systems designed to prevent stagnation, monitored for microbial and endotoxin levels.
- Compressed Gases: Filtered at point-of-use; regularly tested for microbial quality.
- Clean Steam: Validated for sterility and non-pyrogenicity.
5.8 Material and Component Transfer
- Incoming materials are quarantined, cleaned, and transferred through airlocks and material pass boxes.
- Double wrapping and transfer disinfection techniques used.
- Movement between classified areas follows unidirectional and clean-to-dirty flow.
5.9 Aseptic Process Simulation (Media Fill)
- Conducted bi-annually or after changes.
- Simulates actual worst-case interventions and filling conditions.
- Zero tolerance for contaminated units (≤3000 units); per Annex 1 and PDA TR#22.
5.10 Product and Process Monitoring
- Sterility testing, bioburden monitoring, endotoxin testing, and in-process controls form part of the quality assurance program.
- Deviation management and real-time investigations.
6. Quality Risk Management (QRM)
- Risk assessments conducted for all contamination sources using FMEA or HACCP principles.
- Each risk is categorized, mitigated, and monitored.
- Risk acceptance aligned with product criticality and patient safety.
- Conduct detailed Quality Risk Management (QRM) to identify potential contamination risks across the facility, people, materials, and processes.
- Develop and maintain a risk register to prioritize control measures and monitoring efforts according to contamination impact
7. Data Integrity and Documentation
- All activities are documented in real-time using ALCOA+ principles.
- Audit trails maintained for electronic systems.
- EM, sterilization, cleaning, and maintenance records are traceable and reviewable.
8. Continuous Improvement
- CCS is a living document—periodically reviewed and revised based on:
- EM trends
- Deviation investigations
- Regulatory feedback
- Scientific advancements
- EM trends
- CAPAs are implemented for identified gaps with effectiveness checks.
9. Training and Awareness
- CCS is communicated to all relevant staff.
- Regular training sessions ensure understanding of contamination risks and control measures.
- Training effectiveness is assessed through qualification, observation, and audits.
10. Conclusion
The CCS integrates facility, equipment, personnel, materials, and process controls into a cohesive strategy to ensure sterility assurance. This approach complies with global regulatory expectations and is central to patient safety and product quality.